La retinopatía sobre la zona próxima a la mácula y en los Asiaticos mas allá de la mácula, es un efecto adverso importante a tener en cuenta en el tratamiento con antimaláricos. Datos epidemiológicos mencionaban prevalencias no mayores al 3% (1,2), pero con estudios mas modernos que detectan el daño antes de que existan cambios en el fondo de ojo llega a ser del 7.5% (3). La mayoría de la información publicada es de HCQ.
Recomendaciones de la American Academy of Ophthalmology, sobre la detección de la retinopatía por cloroquina (CQ) e hidroxicloroquina (HCQ). (4)
- Patrón de retinopatía: Aunque en la mayoría de casos, el lugar del daño tóxico es parafoveal, los pacientes asiáticos a menudo muestran un patrón de daño extramacular.
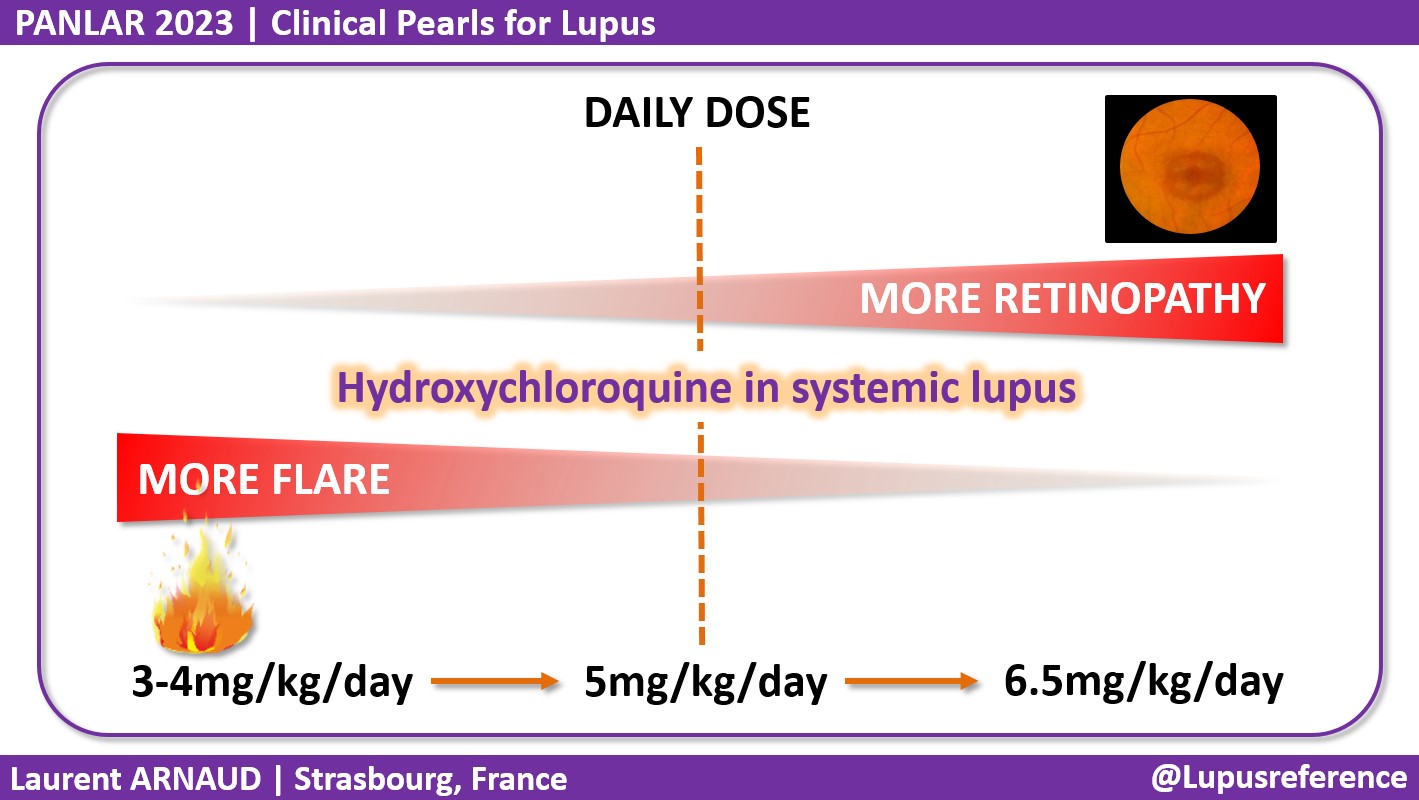
- Dosis: Recomendamos un uso máximo diario de HCQ ≤5,0 mg/kg de peso real, que se correlaciona mejor con el riesgo que el peso ideal. No existen datos demográficos para CQ, pero las comparaciones de dosis en la literatura más antigua sugieren el uso de ≤2,3 mg/kg de peso real.
- Riesgo de toxicidad: El riesgo de toxicidad depende de la dosis diaria y de la duración del uso. Con las dosis recomendadas, el riesgo de toxicidad hasta 5 años es inferior al 1% y hasta 10 años es inferior al 2%, pero aumenta hasta casi el 20% después de 20 años. Sin embargo, incluso después de 20 años, un paciente sin toxicidad tiene solo un 4% de riesgo de conversión en el año siguiente.
- Principales factores de riesgo: Los riesgos más importantes son la dosis alta y el uso prolongado. Otros factores importantes son la enfermedad renal concomitante o el uso de tamoxifeno.
- Programa de detección: Se debe realizar un examen de fondo de ojo inicial para descartar una maculopatía preexistente. Comenzar con la detección anual después de 5 años en pacientes con dosis aceptables y sin factores de riesgo.
- Pruebas de detección: Las pruebas de detección primarias son los campos visuales automatizados más la tomografía de coherencia óptica de dominio espectral (SD OCT). Estas pruebas deben examinar más allá de la mácula central en pacientes asiáticos. El electrorretinograma multifocal (mfERG) puede proporcionar una corroboración objetiva de los campos visuales, y la autofluorescencia del fondo de ojo (FAF) puede mostrar el daño topográficamente. Las pruebas de detección modernas deberían detectar la retinopatía antes de que sea visible en el fondo de ojo.
- Toxicidad: La retinopatía no es reversible y actualmente no existe ningún tratamiento. Es importante reconocerla en una etapa temprana (antes de que se produzca pérdida del epitelio pigmentario de la retina) para prevenir la pérdida de la visión central. Sin embargo, si los resultados de las pruebas no son claros o son cuestionables, deben repetirse o validarse con procedimientos adicionales para evitar la interrupción innecesaria del medicamento (valioso).
- Asesoramiento: Se debe informar a los pacientes (y a los médicos que prescriben) sobre el riesgo de toxicidad, los niveles de dosis adecuados y la importancia de las pruebas de detección anuales periódicas.
Referencias. (Referencia 5 es un documento de lectura recomendado)
1. Mavrikakis I, Sfikakis PP, Mavrikakis E, et al. The incidence of irreversible retinal toxicity in patients treated with hydroxychloroquine: a reappraisal. Ophthalmology. 2003;110(7):1321-1326. doi:10.1016/S0161-6420(03)00409-3
Estudio prospectivo de 15 años (1985-2000), informaron una incidencia general del 0,5% en un grupo de 400 pacientes que recibieron 6,5 mg/kg/día durante al menos seis años y una media de 8,7 años de seguimiento. No se detectó toxicidad en los primeros seis años.
2. Aduriz-Lorenzo PM, Aduriz-Llaneza P, Araiz-Iribarren J, Khamashta MA. Current opinion on hydroxychloroquine-related retinal toxicity screening: where do we stand now?. Lupus. 2020;29(7):671-675. doi:10.1177/0961203320919499
3. Melles RB, Marmor MF. The risk of toxic retinopathy in patients on long-term hydroxychloroquine therapy [published correction appears in JAMA Ophthalmol. 2014 Dec;132(12):1493]. JAMA Ophthalmol. 2014;132(12):1453-1460. doi:10.1001/jamaophthalmol.2014.3459
La prevalencia general de retinopatía por hidroxicloroquina fue del 7,5%, pero esa prevalencia tiene variaciones con el consumo diario (>5 mg/kg) y con la duración del uso (>10 años). Para el consumo diario de 4,0 a 5,0 mg/kg, la prevalencia de toxicidad retiniana se mantuvo por debajo del 2% dentro de los primeros 10 años de uso, pero aumentó a casi el 20% después de 20 años de uso. Otros factores de riesgo importantes incluyen la enfermedad renal y el tratamiento concomitante con citrato de tamoxifeno.
4. Marmor MF, Kellner U, Lai TY, Melles RB, Mieler WF; American Academy of Ophthalmology. Recommendations on Screening for Chloroquine and Hydroxychloroquine Retinopathy (2016 Revision). Ophthalmology. 2016;123(6):1386-1394. doi:10.1016/j.ophtha.2016.01.058.
5. Donate Tercero, A., & Marchite, C. B. Retinopatía por antipalúdicos. Control oftalmológico. Laboratorios Thea Retrieved November 1, 2024