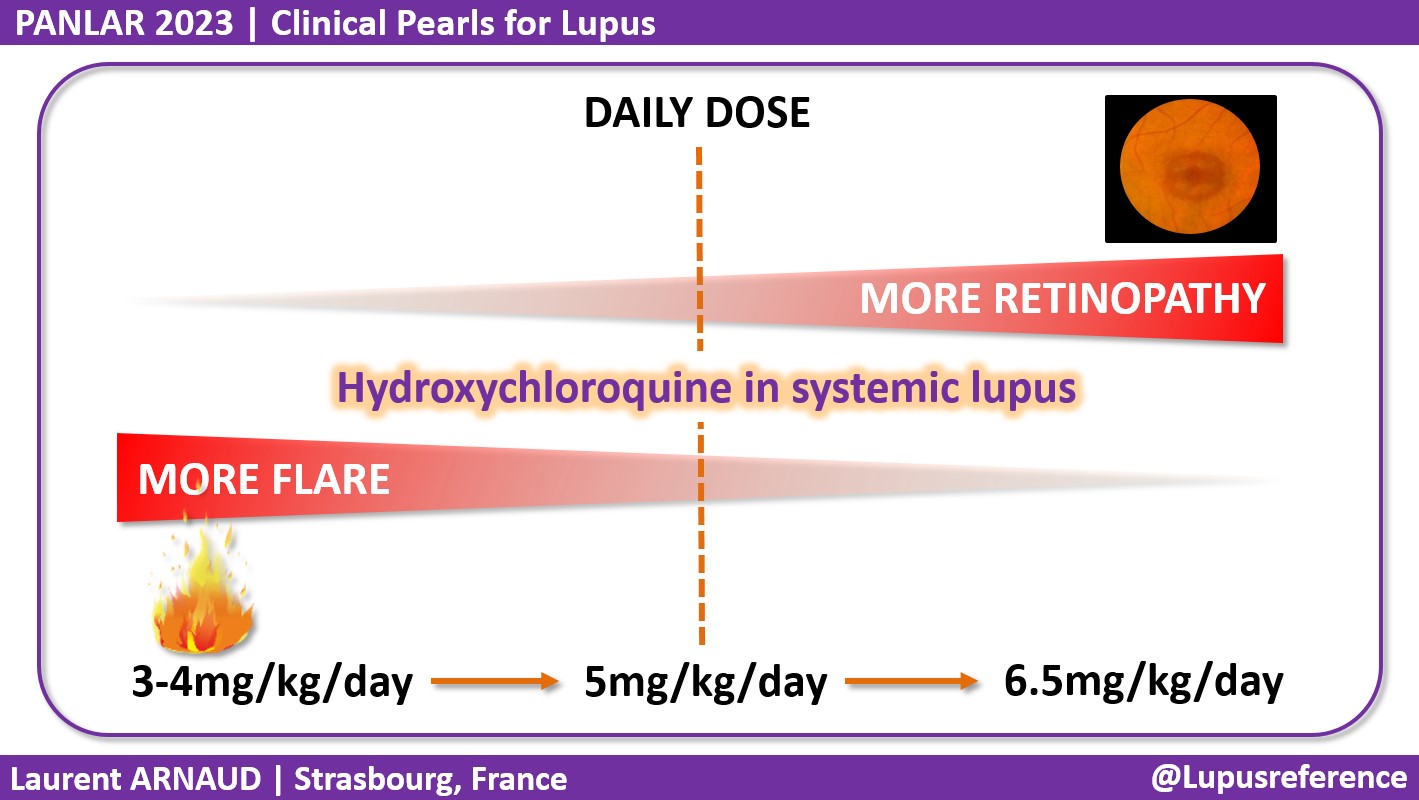
La artropatía de Jaccoud causa deformidades en manos y pies por la inflamación intermitente de los ligamentos y los tejidos blandos periarticulares. Puede verse flexión y subluxación metacarpofalángicas, desviación cubital de los dedos 2 a 5 de la mano, dedos en «cuello de cisne», pulgar en «Z», hallux valgus, subluxaciones metatarsofalángicas y otras subluxaciones no erosivas inicialmente (con el tiempo erosiones diferentes a las observadas en la artritis reumatoide). Estas deformidades son reversibles durante años pero finalmente se hacen fijas. El tratamiento se dirige a controlar la inflamación articular precozmente y prevenir la limitación de la movilidad articular y la pérdida de la función articular.
Esta artropatía deformante se describió inicialmente en pacientes con fiebre reumática crónica y episodios recurrentes de artritis, pero puede aparecer en otras enfermedades. En 1975, Bywaters utilizó el término síndrome de Jaccoud para describir una artropatía similar en pacientes con otras enfermedades reumáticas, como lupus eritematoso sistémico (LES), enfermedad de Sjögren, esclerodermia, dermatomiositis, síndromes de solapamiento, enfermedad mixta del tejido conjuntivo, arteritis de Takayasu, vasculitis necrosantes de mediano vaso, púrpura de Schönlein-Henoch, espondilitis anquilosante, artritis psoriásica y enfermedad por depósito de pirofosfato cálcico.
Actualmente, la mayoría de los pacientes con artropatía de Jaccoud presentan LES, aunque pueden tardar años en cumplir los criterios de la clasificación diagnóstico de LES. No es raro que dichos pacientes sean diagnosticados antes de síndrome semejante al lupus o de conectivopatía indiferenciada. Las manifestaciones del aparato locomotor son muy frecuentes en el LES desde el inicio de la enfermedad, y casi todos los pacientes presentarán síntomas articulares a lo largo de su vida. Las manifestaciones articulares son heterogéneas y van desde simples artralgias hasta una artritis deformante y erosiva. Casi el 70% de los pacientes presentan artritis no deformante, intermitente o persistente (1,2). Entre los pacientes que desarrollan deformidades pueden diferenciarse tres grupos:
- Algunos pacientes presentan una artropatía deformante no erosiva
- Algunos una artropatía erosiva similar a la artritis reumatoide. La artritis erosiva se ha considerado como un solapamiento AR-LES o como una manifestación articular grave del LES y es diferente de la artropatía de Jaccoud.
- Otros una artropatía de Jaccoud con erosiones diferentes a las observadas en la artritis reumatoide.