Cristhyan Pacheco-Ayos
Medicina Interna – Universidad de Cartagena
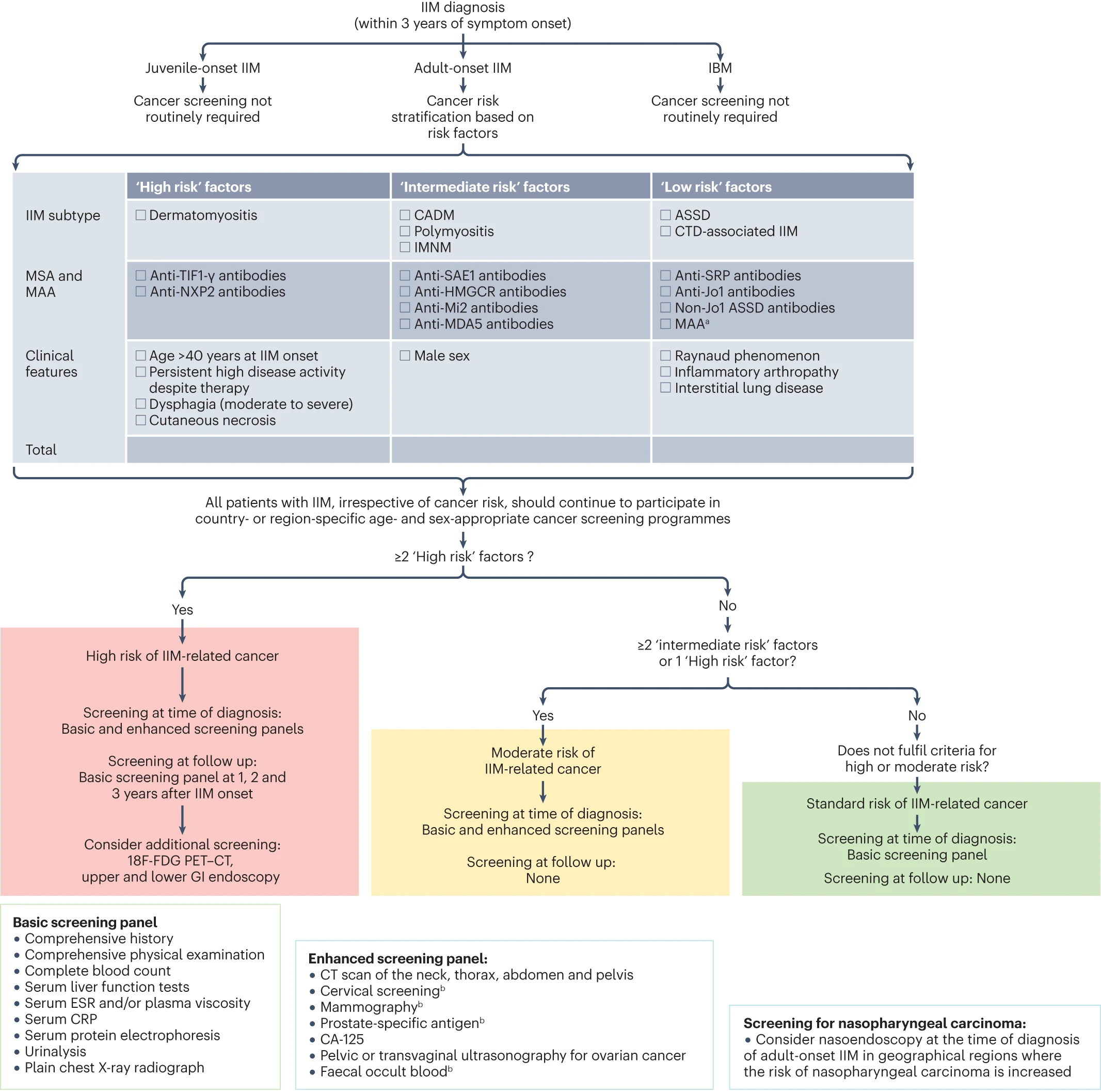
Introducción
La osteoporosis es una enfermedad esquelética
sistémica caracterizada por una baja densidad mineral ósea y un deterioro en la
microarquitectura ósea que confiere un mayor riesgo de fracturas por fragilidad
(1). La prevalencia a nivel mundial se estima alrededor del 19.7% y de baja
masa ósea del 40.4%, siendo esta enfermedad más frecuente en mujeres
posmenopáusicas (un 27.4%) y su prevalencia va aumentando conforme se avanza en
edad, llegando a cifras del 50% a los 70 años y entre un 70-80% a partir de los
80 años (2). A nivel nacional se estima una prevalencia de 2.440 casos por
100.000 habitantes basado en registros hasta el 2018. (3)
Existen múltiples factores de riesgo descritos, dentro
de los cuales destacan el sexo femenino (RR 2.34), tabaquismo (RR 1.14), IMC
<18.5 (2.76%). Asimismo, enfermedades sistémicas como la infección por VIH,
Artritis Reumatoide, Diabetes mellitus, trastornos endocrinos y el uso de
fármacos como glucocorticoides, inhibidores de la bomba de protones, entre
otros (4-5)
El diagnóstico de esta enfermedad se puede realizar en
dos escenarios:
1er escenario: Diagnóstico clínico
Si existe una fractura por fragilidad, las cuales
clínicamente se definen como aquella posterior a un trauma de bajo impacto o
caída de propia altura, siendo las más frecuentes en vertebras dorsolumbares,
cadera, radio distal y humero proximal. Esto se considera un diagnóstico tardío
que confiere mayor riesgo por la mortalidad y riesgo de nuevas fracturas
asociadas (6)
2do escenario: Diagnóstico densitométrico
Siendo una enfermedad silente, surgen tres preguntas: ¿Cuál
prueba se indica?, ¿Cuándo indicamos una prueba?, Y una regla de oro en medicina
que dice “no ordenar una prueba que no sepamos interpretar”, por lo que la
última pregunta es ¿Cómo se interpreta?
El gold standard para diagnóstico de la osteoporosis
es la densitometría ósea por absorción dual de rayos X, el cual se adoptó desde
1994 por la OMS como método de rutina para su detección.
Esta se basa en el principio de cuantificar la
densidad mineral ósea (DMO) en sitios de interés previamente definidos los
cuales son:
Columna lumbar: Desde la vertebra L1 hasta L4 en una proyección posteroanterior
Cadera:
Se puede usar la medición de cadera total o cuello femoral y elegir la de menor
DMO
Radio:
Usar el radio 33% de la mano no dominante
Cada uno de estos sitios de interés tiene aspectos
técnicos en cuanto a la posición del paciente para la toma de la imagen y
ciertas condiciones que deben cumplirse para su adecuada interpretación, las
cuales explicaremos secuencialmente con ejemplos de densitometrías reales.
Columna lumbar
Paso 1) Posición de las vértebras: Estas deben estén centradas y alineadas, dado que
desviaciones escolióticas significativas pueden alterar la DMO medida. En la
imagen obtenida pueden observarse las crestas iliacas, pero estas no deben
estar incluidas en la región de interés. Podemos ver en la vertebra L4 la
región de interés presenta una angulación con respecto a las vértebras
superiores, esto debido a escoliosis presente.
Paso 2) Excluir vértebras anatómicamente anormales: La presencia de cambios artrósicos, fracturas o
patologías propias de la vertebra pueden alterar la DMO y por lo tanto el
diagnóstico densitométrico. En este caso no se excluyó ninguna
Paso 3) Alteraciones de la DMO por causas no-óseas: Calcificaciones de otras estructuras (nefrolitiasis,
ganglios calcificados, ateromatosis aórtica) que se pueden superponer a las
vértebras pueden generar alteración de la DMO, asimismo la presencia de
artefactos metálicos como material de osteosíntesis pueden generar el mismo
error. No se observan ninguna de estas alteraciones
Paso 4) Diferencias mayor a 1 desviación estándar (DE)
entre vertebras contiguas: Como
se observa en la tabla amarilla, se reporta el T score de cada vertebra: -0.8,
-1.2, -2.1 y -1.6 desde L1 a L4 respectivamente. Entre vértebras contiguas
(L1-L2, L2-L3 y L3-L4) no existe una diferencia mayor a 1 DE por lo cual se
pueden usar todas para el promedio final
Paso 5) Número de vértebras para interpretación: Deben quedar al menos 2 vértebras contiguas (L1-L2,
L2-L3 o L3-L4) para poder considerarse la columna lumbar como sitio
interpretable para el diagnóstico densitométrico. En este caso se cumple lo
anterior por lo cual se considera interpretable
Cadera
Paso 1) Posición de la cadera: La posición del fémur al momento de realizar la DXA puede
afectar la DMO, el tecnólogo debe asegurarse de que la cadera esté en rotación
interna (con aproximadamente 15 -25 ° de rotación). Para saber si esta posición
es correcta el trocánter menor (señalado con la flecha) no debe ser visible. En
este caso sí lo es, por lo que se concluye un mal posicionamiento de la cadera
y puede llevar a errores en la medición de la DMO
Paso 2) Región de interés: En cadera se tienen los siguientes sitios de medición:
cuello, trocánter, triangulo de Ward y cadera total. Se recomiendan de elección
cuello y cadera total (puede ser derecho o izquierdo, cual sea más bajo)
Paso 3) Alteraciones de la DMO: La presencia de grasa abdominal abundante sobre esta
región puede generar alteración en la lectura, en caso de que la haya se
recomienda se retire para la toma de imágenes. Al igual que en columna lumbar,
la presencia de osteoartrosis grave o artefactos metálicos (pines, tornillos o
en su defecto prótesis total) puede generar lecturas alteradas, por lo que se
recomienda elegir la cadera que no tenga estos artefactos.
Antebrazo
Paso 1) Antebrazo de elección: Elegir el antebrazo no dominante del paciente
Paso 2) Posición: Debe estar alineado con el eje longitudinal del
escáner
Paso 3) Artefactos: Debe excluirse si hay presencia de material de
osteosíntesis
Paso 4) Región de interés: Para el antebrazo se tomará en cuenta la zona
denominada radio 33
Paso 5) Pertinencia: El antebrazo solo se elegirá como interpretable cuando
no se puedan interpretar la columna lumbar o cadera. Puede usarse
adicionalmente en casos de osteoporosis secundaria a hiperparatiroidismo
conocido. O cuando el peso del paciente no permita su colocación en la camilla
para la toma de columna o cadera.
¿Qué hago con los valores obtenidos?
La gráfica anterior nos muestra la campana de Gauss
que corresponde a una distribución normal de la población, siendo el referente
de normalidad la DMO de una mujer blanca entre 20-29 años según la ISCD.
Los puntos de corte definidos por la OMS para
interpretación son los siguientes
|
|
T score
|
Z score
|
|
Normal
|
≥-1 DE
|
> -2 DE
|
|
Baja masa ósea
|
<-1 DE y > -2.5
DE
|
≤ -2 DE
|
|
Osteoporosis
|
≤ -2.5 DE
|
*No aplica*
|
Cuando comparamos el resultado de nuestro paciente con
el estándar, a este valor lo llamaremos T score cuyos valores
corresponde a cuantas desviaciones estándar se acerca o aleja de la normalidad.
Si comparamos a nuestro paciente con alguien de su misma edad, sexo y raza,
este valor se llamará Z score.
¿Cómo interpreto los resultados?
Paso 1) El
diagnóstico de osteoporosis se debe hacer solo con los puntos de corte del T
score. Para el Z score se hablará de “baja masa ósea para la edad”. Esta medida
es más útil en niños o adultos menores de 50 años
Paso 2) Evaluar
los sitios de interés en columna lumbar, cadera y/o antebrazo según se explicó
anteriormente para evaluar si son interpretables
Paso 3)
Elegir el peor valor posible, es decir: Si en columna lumbar se documenta baja
masa ósea, pero en cadera hay osteoporosis, el diagnóstico densitométrico será
osteoporosis.
Paso 4)
Si tanto en cadera como columna lumbar se reportan como normales/baja masa
ósea, pero en antebrazo hay osteoporosis, se debe interpretar según el valor
más bajo de columna o cadera. Antebrazo solo se debe tener en cuenta en casos
de hiperparatiroidismo o si los dos anteriores no son interpretables
Paso 5) Si
se diagnosticó osteoporosis, se debe establecer la gravedad y evaluar la
presencia de fracturas vertebrales asintomáticas para definir el mejor
tratamiento posible
Paso 6) Si
se diagnosticó baja masa ósea, se debe establecer el riesgo de fractura con la
herramienta FRAX validada para la población del paciente y evaluar si se
beneficia de tratamiento a pesar de no tener un diagnóstico densitométrico de
osteoporosis.
Paso 7) Si
el estudio es normal, se debe anotar la fecha de realización para la evaluación
periódica según la indicación clínica o de tamización y hacer las comparaciones
posteriores.
Conclusiones
La densitometría ósea es un examen accesible en nuestro país y de fácil interpretación una vez conocidas y entendidas las bases para su interpretación que puede realizar cualquier profesional de la salud para lograr diagnósticos y tratamientos tempranos con el fin de prevenir las fracturas por fragilidad y su morbimortalidad asociada.
Referencias
1. Consensus development conference: diagnosis, prophylaxis, and treatment of osteoporosis. Am J Med, 1993. 94(6): p. 646-50
2. Fernández-Ávila DG, Bernal-Macías S, Parra MJ, Rincón DN, Gutiérrez JM, Rosselli D. Prevalence of osteoporosis in Colombia: Data from the National Health Registry from 2012 to 2018. Reumatol Clin (Engl Ed). 2021;17:570–4,
3. Panday, K., A. Gona, and M.B. Humphrey, Medication-induced osteoporosis: screening and treatment strategies. Ther Adv Musculoskelet Dis, 2014. 6(5): p. 185-202.
4. Amarasekara, D.S., J. Yu, and J. Rho, Bone Loss Triggered by the Cytokine Network in Inflammatory Autoimmune Diseases. J Immunol Res, 2015. 2015: p. 832127
5. J.A. Kanis, L.J. Melton 3rd, C. Christiansen, C.C. Johnston, N. Khaltaev. The diagnosis of osteoporosis. J Bone Miner Res., 9 (1994), pp. 1137-1141
6. Krueger D, Tanner SB, Szalat A, Malabanan A, Prout T, Lau A, Rosen HN, Shuhart C. DXA Reporting Updates: 2023 Official Positions of the International Society for Clinical Densitometry. J Clin Densitom. 2023 Nov 2;27(1):101437. doi: 10.1016/j.jocd.2023.101437
7. Martineau P, Morgan SL, Leslie WD. Bone Mineral Densitometry Reporting : Pearls and Pitfalls Bone Mineral Densitometry Reporting : Pearls and Pitfalls. Can Assoc Radiol J. 2021 Aug;72(3):490-504. doi: 10.1177/0846537120919627.
8. José Fernando Molina Restrepo y Luis Alonso González Naranjo. Osteoporosis: enfoque clínico y de laboratorio. Universidad de Antioquia. 2010. Link: https://www.medigraphic.com/pdfs/medlab/myl-2010/myl103-4b.pdf